Students can Download Chapter 1 The Solid State Notes, Plus Two Chemistry Notes helps you to revise the complete Kerala State Syllabus and score more marks in your examinations.
Kerala Plus Two Chemistry Notes Chapter 1 The Solid State
General Characteristics of Solid State
Definite mass, volume and shape; short inter molecular distances; strong intermolecular forces; constituent particles have fixed position and can only oscillate about their mean positions; incompressible and rigid.
Classification of Solids:
1. Crystalline Solids :
The constituent particles are well orderly arranged. It has long range orcfe/-which means that there is a regular pattern of arrangement of particles which repeats itself periodically overthe entire crystal, e.g. NaCl, Quartz.
2. Amorphous Solids (Greek amorphos = no form):
The constituent particles are irregularly arranged; i.e. they have irregular shape, the arrangement of constituent particles in this solid has only short range order, e.g. rubber, quartz, glass, plastic.
Amorphous solids have a tendency to flow slowly, therefore these are called pseudo solids or supercooled liquids, e.g. Glass – Glass flows down very slowly and makes the bottom portion slightly thicker.
Crystalline solids are anisotropic in nature (i.e. their physical properties like electrical resistance or refractive index show different values in different directions). Amorphous solids on the other hand are isotropic, (i.e. their physical properties would be same along any direction)
Difference between Crystalline and Amorphous Solids

Classification of Crystalline Solids:
Crystalline solids can be classified on the basis of nature of intermolecular forces operating in them into four categories.
![]()
Molecular Solids :
In this, molecules are the constituent particles, they are of three categories:
a. Non-polar Molecular Solids :
They are formed by the regular arrangement of either atoms or non-polar molecules held by weak dispersion forces or London forces. They have low melting points and are liquids or gases at room temperature and pressure, e.g. solid forms of argon, He, H2 and Cl2, l2.
b. Polar Molecular Solids :
In these polar molecules are held by relatively stronger dipole-dipole interaction. They are soft and non-conductors of electricity e.g. solid forms of HCl, SO2, NH3.
c. Hydrogen bonded Molecular Solids :
Strong H- bonding binds the molecules of these solids. They are non-conductors of electricity and are volatile liquids or soft solids.
e.g. Ice (Solid H2O)
Ionic Solids :
In these ions are held together by strong coulombic (electrostatic) forces. They are hard and brittle in nature and have high melting point and boiling points. They are electrical insulators in the solid stale, but in molten state/solutions conduct electricity because ions become free to move.
e.g. NaCl, KCl, KNO3 etc.
Metallic Solids :
In these positive ions are surrounded by and held together by a sea of free electrons. These free and mobile electrons are responsible for high electrical and thermal conductivity. Due to this mobile electrons metals have lustre and colour, e.g. Gold, Silver etc.
Covalent or Network Solids:
They have covalent bonds between adjacent non-metal atoms which are held strongly in their positions. They have high melting points and are insulators, e.g. Diamond, SiC Graphite is an exceptional case – it is soft and conducts electricity due to its typical layer structure.
Crystal Lattice and Unit Cells
The three dimensional arrangement of constituent particles in a crystal, represented by points is called crystal lattice/space lattice.
Unit Cell:
The smallest repeating portion of space lattice/crystal lattice. A unit cell is characterised by,
1. The distance along with three edges: a, b & c
2. Angle between the edges:
α (between b & c)
β (between a & c)
γ (between a & b)
Primitive and Centred Unit Cells
a. Primitive Unit Cell :
the constituent particles are present only on the corner positions of unit cell.
b. Centred Unit Cells
i. Body-Centred Unit Cell (bcc) :
The constituent particles at all the corners as well as at the centre of the unit cell.
ii. Face-Centred Unit Cell (fee) :
The constituent particles at all the corners as well as at the centre of the each face.
iii. End-Centred Unit Cell:
One constituent particle is present at the centre of any two opposite faces besides the one present at its corners.

Seven Primitive Unit Cells and their Possible Variations as Centred Unit Cells:

Number of Atoms in a Unit Cell
1. Primitive Cubic Unit Cell or Simple Cubic Unit Cell:
Primitive cubic unit cell has atoms only at its corner. Each atoms at a corner is shared between eight adjacent unit cells. Therefore the contribution of an atom/molecule to a particular unit cell is 1/8

Total number of atoms in unit cell = 8 × 1/8 = 1
![]()
2. Body Centred Cubic Unit Cell (bcc):
In bcc unit cell, particles are present not only at the eight corners but also at the centre of the cube.

In a bcc unit cell:
i. 8 corners x 1/8 per corner atom = 8 × 1/8 = 1 atom
ii. 1 body centre atom = 1×1 = 1 atom
∴ Total number of atoms per unit cell = 1 + 1=2 atoms
3. Face Centred Cubic Unit Cell :
A fee unit cell contains atoms at all the corners as well as at the centre of 6 faces of the cube. The atoms at the face centre is shared between two adjacent unit cells and the contribution is only ½ to the unit cell.

In a fcc unit cell:
i. 8 comers x 1/8 per corner atom = 8 × 1/8 = 1 atom
ii. 6 face centre x ½ atom per unit cell = 6 × ½ = 3 atoms
∴ Total number of atoms per unit cell = 1+3 = 4 atoms
Close Packed Structures:
In solids, the constituent particles (considered as identical hard spheres) are close-packed, leaving the minimum vaccnt space.
Coordination number (C.N.) :
The number of nearest neighbouring particles (spheres) in a crystal lattice is called coordination number.
a. Close Packing in One Dimension :
In this arrangment, each sphere is in contact with two of its neighbours. The cordination number is two.
![]()
b. Close Packing in Two Dimensions :
Two dimensional close packing can be done in two different ways:
i. Square Close Packing (scp):
The second row is placed in contact with the first one such that the spheres are exactly above those of the first row. The spheres of the two rows are aligned horizontally as well as vertically. Each sphere is in contact with four of its neighbours. Thus coordination number is 4. This is also known as AAA type arrangment.

ii. Hexagonal Close Packing (hep) :
The spheres in every second row are placed in the depressions of the first row. The spheres in the third row are seated in the depressions of the second row and so on. The fourth layer is aligned with the second layer. Hence this type of arrangement is of ABAB type. Each sphere is in contact with 6 neighbouring spheres. Thus the C.N. is 6.
In hexagonal close packing, particles are more closely packed than in square close packing. Hence, it is more efficient than square close packing.
![]()
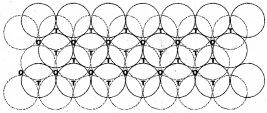
c. Close Packing in Three Dimensions :
i. Three-dimensional close packing from two-dimensional square close-packed layers :
the second layer is placed over the first layer such that the spheres of the upper layers are exactly above those of the first layer. Here, spheres of both the layers are perfectly aligned horizontally as well as vertically. This type of arrangement has AAA type pattern. This arrangement generates the simple cubic lattice.

ii. Three dimensional close packing from two-dimensional hexagonal close-packed layers
a. Placing second layer over the first layer :
The spheres of the second layer are placed in the depressions of the first 2 dimensional hep layer. Wherever a sphere of the second layer is above the triangular void of the first layer (or vice versa) a tetrahedral void is formed.|
The vaccant space/sites in close packed structure are called voids/interstitial voids.
Tetrahedral void (td void) :
The vacant space surrounded by 4 spheres are called tetrahedral voids.

Octahedral void (oh void):
The vaccant space surrounded by six spheres in a close packed arrangement.

If ‘N’ is the number of spheres in close packed structure,
b. Placing third layer over the second layer :
If particles in the third layer are arranger) in the tetrahedral voids, the spheres of the third layer are exactly aligned with those of the first layer. Thus, the pattern of spheres is repeated in alternate layers. This is known as ABAB or hexagonal close packed (hep) structure, e.g. Mg, Zn, Cd etc.

If particles in the third layer are arranged in the octahedral voids, spheres of the fourth layer are aligned with those of the first layer. Thus, ABCABC … arrangement is obtained. This is known as cubic close packed (ccp) structure or face-centred cubic (fee) structure, e.g. Cu, Ag, Au etc.

Both the hep and ccp are highly efficient and 74% space in the crystal is filled. Coordination number is 12 in either of these two structures.

Packing Efficiency
It is the percentage of total space filled by the particles.
Let ‘a’ be the edge length of a unit cell and ‘r’ the radius of sphere.
Packing Efficiency in ccp and hep Structures:
In the case of ccp and hep, the edge length,

Packing Efficiency of Body Centred Cubic Structures :
In this case radius of a sphere,

Packing Efficiency in Simple Cubic Lattice :
In simple cubic lattice edge length, ‘a’ and radius of the sphere ‘r’ are related as,
a = 2r
r = a/2
We know that a simple cubic unit cell contains only one sphere.

Calculations Involving Unit Cell Dimensions :
Edge length of unit cell = a
Volume of the unit cell = a³
Mass of unit cell = Number of atoms in unit cell × mass of each atom = z × m
Mass of an atom in unit cell m = \(\frac{M}{N_{A}}\) (M-molar mass)

Note:
The density of the unit cell is the same as the density of the substance.
Imperfection in Solids :
The crystal defects are irregularities in the arrangement of constituent particles. There are two types of defects:
1. Point Defects:-
Irregularities from ideal arrangement around a point or an atom.
2. Line Defects:-
Irregularities/deviations from ideal arrangement in entire rows of lattice points.
![]()
Point Defects:
They can be classified into three types – stoichiometric defects, impurity defects and non-stoichiometric defects.
a. Stoichiometric Defects:
These are the point defects that do not disturb the stoichiometry of the solid. They are also called intrinsic or thermodynamic defects. These are of two types:
i. Vacancy Defect:
When some of the lattice sites are vacant (missing of constituent particles), the crystal is said to have vacancy defect. As a result density of the substance decreases.
ii. Interstitial Defect:
When some constituent particles occupy an interstitial site, the crystal is said to have interstitial defect.
There are two types of stoichiometric defects in ionic solids : Schottky Defect and Frenkel Defect.
Schottky Defect:
It arises due to the missing of equal number of cations and anions from their normal positions leaving behind a pair of holes. It is observed in ionic compounds having high coordination numberwith ions of almost similar size. Since equal number of cations and anions are missing they maintain electrical neutrality. Density of the substance decreases.
e.g. NaCl, KCl, CsCl and AgBr.
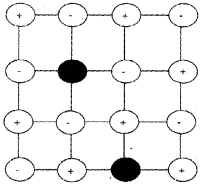
Frenkel Defect:
It arises due to an ion, usually cation which is dislocated from its normal site to an interstitial site. It creates a vaccancy defect at its original site and an interstitial defect at its new location. This is also called dislocation defect. It is usually observed in ionic compounds having low coordination number and crystals with anions much larger in size than the cations. Since no ions are missing, density of the solid does not change, e.g. ZnS, AgCI, AgBrandAgl.
Note: AgBr shows both Schottky as well as Frenkel defects.

Note: AgBr shows both Schottky as well as Frenkel defects.
b. Impurity Defects :
Defect caused by foreign ions. e.g. If molten NaCl containing a little amount of SrCl2 is crystallised, some of the sites of Na+ ions are occupied by Sr2+. Each Sr2+ replaces two Na+ ions. It occupies the site of one Na+ ion and the othersite remains vaccant. The cationic vaccancies thus pro duced are equal in number to that of Sr2+ ions. Another example is the solid solution of CdCl2 and AgCl.

c. Non-Stoichiometric Defects:
The stoichiometry of the crystal is altered due to defects. These defects are of two types:
i. Metal Excess Defect
ii. Metal Deficiency Defect
![]()
i. Metal Excess Defect
1. Due to anionic vacancies :
A compound may have excess of metal ion if a negative ion is absent from its lattice site leaving a hole which is occupied by electron. The anionic sites occupied by unpaired electrons are called F-centres. (German word Farbenzenter means colour centre). These can impart colourto the crystal by excitation of electrons when they absorb energy from visible light, e.g. When crystals of NaCl are heated in an atmosphere of Na vapour, F-centres are formed. As a result, the crystal has an excess of sodium which imparts yellow colour.
Excess Li in LiCl – imparts pink colour
Excess K in KCl – imparts violet(lilac) colour

2. Metal excess defect due to extra cations at interstitial sites :
Here, electrical neutrality is maintained by the presence of an electron in the interstitial site. e.g. Zinc oxide (ZnO) is white in colour at room temperature. On heating it loses O2 and turns yellow.
![]()
Now there is excess of zinc in the crystal and its formula becomes Zn1+x O. The excessZn2+ ions move to interstitial sites and electrons move to neighbouring interstitial sites.
ii. Metal Deficiency Defect
1. Due to cation vaccancies:
A positive ion is missing from its lattice postion and the extra negative charge thus created is balanced by the adjacent metal ion which attains one additional positive charge.
e.g. FeO. It is mostly found with a composition of Fe0.95O. In crystal of FeO some Fe2+ cations are missing and the loss of positive charge is made up by the presence of required number of Fe3+ ions. As a result, the crystal has metallic lustre. Other example is FeS (fool’s gold).
2. Due to the presence of anions in interstitial site:
An extra negative ion would occupy an interstitial site and the extra negative charge thus formed is balanced by an adjacent cation possessing additonal positive change. This defect is not common because anions are bigger than cations and cannot be occupied in interstitial sites.
Properties of Solids
Electrical Properties :
Solids can be classified into 3 types on the basis of their conductivities.
1. Conductors :
Solids with conductivities of the order of 104 to 107 ohm-1 m-1. Metals are good conductors of electricity and the conductivity is in the order of 107 ohm-1 m-1.
ii. Insulators :
The solids which almost do not allow the passage of electricity, e.g. S, P, wood, paper, rubber. Conductivity order 10-20– 10-10 ohm-1 m-1
iii. Semiconductors :
The solids whose conductivity lies between metallic conductors and insulators. Conductivity range from 10-6 to 104 ohm-1 m-1 (or 10-8 to 102 ohm-1cm-1).
Oxides like TiO, VO, ReO3 etc. are good conductors. But oxides like Ti2O3, V2O3 etc. behave as insulators at centain termperature. TiO2, V2O5 etc. are perfect insulators. The conductivity of semiconductors increases with temperature while that of metals decreases with temperature.
Conduction of Electricity in Metals :
Metals conduct electricity in solid as well as molten state through movement of electrons. The conductivity of metals depend upon the number of valence electrons available per atom. The atomic orbitals of metal atoms form molecular orbitals which are so close in energy to each other as to form a band. If this band is partially filled or if overlap with a higher energy unoccupied conduction band, then the electrons can flow easily under an applied electric field.

Conduction of Electricity in Semiconductors :
Here, the energy gap between the valence band and conduction band is small. Therefore some electrons may jump to conduction band and show some conductivity. Electrical conductivity of semi conductors increases with rise in temperature, since more electrons can jump to conduction band. This type of semiconductors are known as intrinsic semiconductors, e.g. Silicon, Germanium.
The conductivity of intrinsic semi conductors is increased by adding appropriate amount of suitable impurities, which introduce electronic defects. This process is called dopping. It can be done in two ways:
a. By adding electron rich impurities (n-type semiconductors) :
If Si and Ge are dopped with group 15 elements like P or As, the 5th electron is extra and becomes delocalised which is responsible for electrical conductivity. Since conductivity is due to the negatively charged electron it is called n-type semiconductor.
![]()
b. By adding electron deficit impurities (p-type semi conductors):
If Si and Ge are dopped with group 13 elements like B, Al or Ga an electron deficient bond or electron hole is produced. Under the influence of electric field, electrons move towards the positively charged plate through electronic holes. It appears as if electron holes are positively charged and are moving towards negatively charged plate. This type semi conductors are called p-type semi conductors.
Application of n-type and p-type semi conductors:
- Diode is a combination of n-type and p-type semi conductors and is used as a rectifier.
- The npn and pnp types of transistors are used to amplify radio or audio signals.
- Photo-diode is used for conversion of light energy into electrical energy.
Magnetic Properties :
Every substance has some magnetic properties associated with it. Each electron in an atom behaves like a tiny magnet. Its magnetic moment originates from two types of motions.
i. Electron’s orbital motion around the nucleus
ii. Electron’s spin around its own axis
Magnetic moment is measured in Bohr magneton
(B.M), µB. 1 B.M. = 9.27 × 10-24 A m2
Magnetic properties are of 5 types:
i. Paramagnetism :
It is due to the presence of one or more unpaired electrons. Paramagnetic substances are weakly attracted by a magnetic field.
e.g. O2, Cu2+, Fe3+, Cr3+ Ni2+, VO, VO2, CuO, NO
ii. Diamagnetism :
It is due to paired electrons in the substance. Diamagnetic substances are weakly repelled by a magnetic field.
e.g. H2O, NaCl, C6H6, TiO2, N2
iii. Ferromagnetism :
It is considered as an extreme case of paramagentism and is caused by spontaneous alignment of magnetic domains (metal ions grouped into small regions) in the direction of the magnetic field. Ferromagnetic substances are strongly attracted by magnetic field. They retain a permanent magnetism even when the field is removed, e.g. Fe, Co, Ni, Alloys of Fe, Co and Ni, CrO2 Once such a material is magnetised, it remains permanently magnetised.
Alignment of magnetic moments: ↑↑↑↑↑↑
iv. Antiferromagnetism:
It arises due to the alignment of magnetic domains in opposite direction and the resulting moment is zero. Antiferromagnetic substances are expected to possess paramagnetism or ferromagnetism on the basis of unpaired electrons but actually they possess zero net magnetic moment.
e.g. MnO, MnO2, Mn2O3, FeO, NiO, CuO
Alignment of magnetic moments: ↑↓↑↓↑↓
v. Ferrimagnetism :
It is due to the alignment of magnetic moments in opposite directions in unequal numbers resulting in a net magnetic moment. These substances are expected to possess large magnetism on the basis of unpaired electrons but actually have small net magnetic moment and are weakly attracted by magnetic field as compared to ferromagnetic substances.
e.g. Fe3O4 (magnetite), MgFe2O4 & ZnFe2O4.
Alignment of magnetic moments: ↑↑↓↑↑↓